Parallelization of electrostatic over multiple GPUs in GROMACS
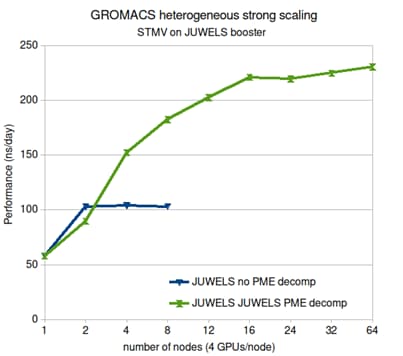
In most molecular dynamics simulations the 3D fast Fourier transform used for calculating the long-range electrostatic interactions is what limits parallelization. The molecular simulation package GROMACS only used to have an efficient parallel implementation for CPUs. Now we have, in a co-design effort with Nvidia, made a parallel electrostatics implementation on GPUs, which also use a new, more efficient spatial decomposition. The implementation is especially beneficial for systems which have direct, fast interconnects between GPUs, as is becoming more common. Being able to use multiple GPU for electrostatics, while dedication other GPUs to the rest of the calculations, has more than doubled the performance of GROMACS on GPU-heavy nodes.
Publication in preparation


