Molecular Highlight: Accurate predictions of GPCR-drug complexes
SeRC researchers have developed methods that enable accurate prediction of receptor-drug interactions at the atomic level.
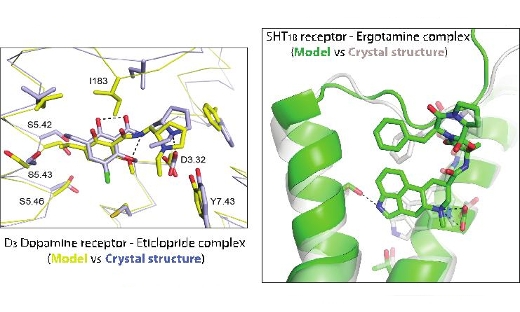
G protein-coupled receptors (GPCRs) are responsible for a large fraction of cellular signaling processes and are the targets of nearly 30% of all marketed drugs. Molecular level understanding of drug binding to GPCRs has been hampered by the lack of experimental high-resolution structures. If accurate computational models of GPCR-drug complexes could be generated, structure-based methods would have the potential to make major contributions to the development of novel pharmaceuticals as tools for ligand discovery and lead optimisation. SeRC researchers have developed methods for atomic-level modeling of GPCRs and have in particular focused on binding site interactions with drugs. Our approach for GPCR structure prediction has been evaluated twice in community-wide assessments (GPCR Dock 2010 and 2013). In both these assessments our modeling strategy has been ranked as one of the best in the field. The blind predictions of the dopamine and serotonin receptors in complex with drugs have been strikingly close to the experimental result (See figure).
References
Carlsson, J.; Coleman, R. G.; Setola, V.; Irwin, J. J.; Fan, H.; Schlessinger, A.; Sali, A.; Roth, B. L.; Shoichet, B. K. (2011) “Ligand discovery from a dopamine D3 receptor homology model and crystal structure.” Nat. Chem. Biol. 7 (11), 769-78.
Kufareva, I.; Rueda, M.; Katritch, V.; GPCR Dock 2010 participants (Carlsson J) ; Stevens, R. C.; Abagyan, R. Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 2011, 19 (8), 1108-26.
Kufareva, I.; Katritch, V.; GPCR Dock 2013 Participants (Rodríguez, R; Ranganathan, R; Carlsson J); Stevens, R. C.; Abagyan, R. Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: meeting new challenges. Structure 2014, In Press.


