VeloxChem: Enabling Quantum Molecular Modeling in High-Performance Computing Environments
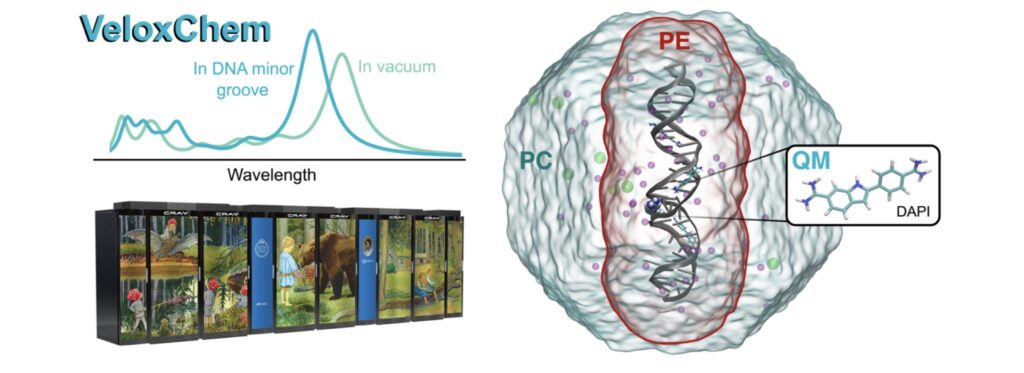
An open-source program named VeloxChem has been developed for the calculation of electronic real and complex linear response functions at the levels of Hartree–Fock and Kohn– Sham density functional theories. With an object-oriented program structure written in a Python/C++ layered fashion, VeloxChem enables time-efficient prototyping of novel scientific approaches without sacrificing computational efficiency, so that molecular systems involving up to and beyond 500 second-row atoms (or some 10,000 contracted and in part diffuse Gaussian basis functions) can be routinely addressed. The underlying hybrid message passing interface (MPI)/open multiprocessing (OpenMP) parallelization scheme makes VeloxChem suitable for execution in high-performance computing cluster environments, showing even slightly beyond linear scaling for the Fock matrix construction with use of up to 16,384 central processing unit (CPU) cores on the Beskow supercomputer at PDC Center for High- Performance Computing. This program development is tightly connected with the EU Horizon 2020 Innovative Training Network titled “Computational Spectroscopy in Natural Sciences and Engineering (COSINE)” (Grant No. 765739).
Image Reference: VeloxChem: A Python-driven density-functional theory program for spectroscopy simulations in high-performance computing environments. Rinkevicius, Z., Li, X., Vahtras, O., Ahmadzadeh, K., Brand, M., Ringholm, M., List, N. H., Scheurer, M., Scott, M., Dreuw, A., Norman, P. WIREs Computational Molecular Science, e1457, 1–14, (2019). https://doi.org/10.1002/wcms.1457


