Software development for exploration and design of complex molecular systems
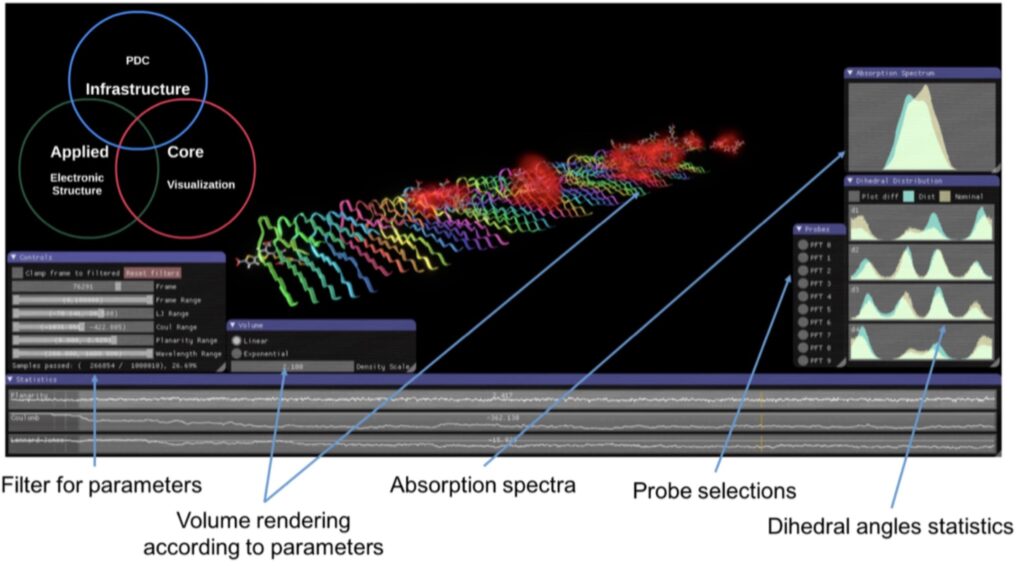
The dominating software for quantum molecular simulations is an American commercial product (Gaussian). The molecular-based software is designed for desktop servers and does not scale beyond a handful of computer nodes, a fact that makes PDC unable to offer software services in quantum molecular science. In an undertaking together with PDC, we will develop a full-fledged DFT program with all the standard capabilities as well as non-standard functionalities developed in the Scandinavian Dalton program community and which provides state-of-the-art scaling on contemporary and future HPC hardware platforms based on Intel, ARM, and Power CPUs as well as NVIDIA GPUs. Moreover, extremely efficient quantum- mechanical (QM) spectral calculations with GP-GPU accelerated time-dependent tight-binding density functional theory (TD-TB-DFT) will make it realistic to envisage real-time user-driven spectroscopy for the exploration and design of complex molecular systems in areas such as drug and probe design, protein and DNA science and technology, and organic electronics. The user interface is provided by the VIAMD software that is developed as a collaborative SeRC project and which presently allows for real-time molecular structure analysis of very large MD data sets. Next, VIAMD will allow for real time analysis also of the electronic structure and its time- dependent responses to external electromagnetic fields as depicted in the Figure.
Fig: VIAMD, a visualization tool developed by researchers in this project proposal enabling analysis by means of filtering through different parameters relating to molecular conformations (today) or time-dependent electronic structure and spectral functions (to be developed).


