Molecular Highlight: The world’s fastest software for Molecular Dynamics on CPUs & GPUs
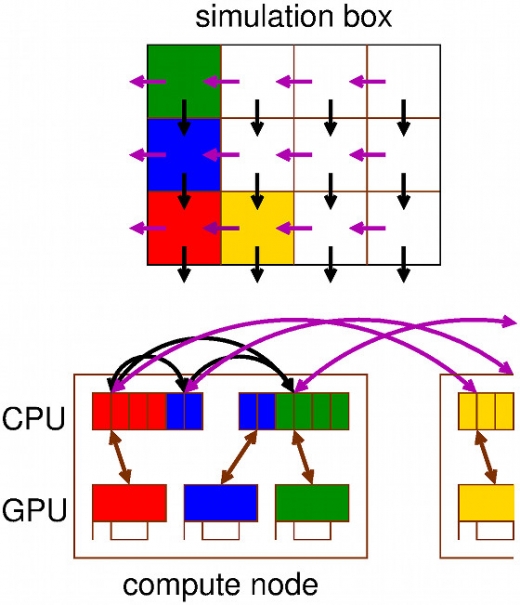
The increase in computing power allows part of (bio) molecular experimental work to be replaced by simulations. However, designing software that leverages the full power of modern (super) computing is becoming more and more challenging due to the fact that hardware is continuously becoming more parallel and heterogeneous.
With molecular dynamics molecular processes can be followed in minute detail, something which is difficult to achieve with experimental techniques, but obtaining this information comes at a high computational cost. The equations of motion of hundreds of thousands of atoms need to be integrated for billions of time steps. As a result, simulation software needs to run highly parallel to make optimal use of modern (super) computers. This is a main goal for the GROMACS molecular simulation package that was developed and is maintained by SeRC researchers. For GROMACS, we have designed multi-level parallelisation, with SIMD (single-instruction multiple-data) parallelisation at the instruction level, OpenMP between cores within nodes and MPI between nodes. We also introduced hybrid acceleration, where the most compute intensive part of the calculation runs on GPUs and the rest on CPUs. GROMACS was already the fastest single CPU code in the world, but is also probably now the fastest parallel and GPU code. Over the course of the lifetime of SeRC, parallelisation has improved by a factor of 4, which together with the improvement of the hardware results in a 10 times shorter time to solution, or conversely 10 times longer simulations. A typical membrane protein system of 300,000 atoms can now scale to 2000 cores or 200 GPUs. GROMACS has become one of the most widely used biomolecular simulation codes in the world, and possibly the most widely spread research software developed in Sweden, with thousands of resulting citations and hundreds of high-impact applications. Since GROMACS is open source software, improvements made possible by SeRC directly benefit thousands of users in academia and industry.
References
Pronk S, Pall S et al. (2013) “GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit” Bioinformatics, 29, 845
Pall S and Hess B (2013) “A flexible algorithm for calculating pair interactions on SIMD architectures” Comput. Phys. Commun. 184, 2641-2650
Pall S, Abraham MJ, Kutzner C, Hess B, Lindahl E (2014) “Tackling exascale software challenges in molecular dynamics simulations with GROMACS” Proc. EASC2014 (in press)


