Electronic Highlight: On the Influence of Water on the Electronic Structure of Firefly Oxyluciferin Anions
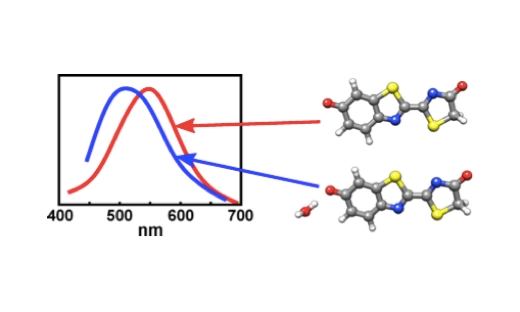
Combining molecular dynamics simulations and time-dependent density functional theory calculations indicate that the preferred binding site for the water molecule is the phenolate oxygen of the anion.
Here, action spectroscopy has been used to compare the absorption by the firefly oxyluciferin lumophore isolated in vacuo and complexed with a single water molecule. Combined molecular dynamics simulations and time-dependent density functional theory calculations closely reproduce the broad experimental peaks and also indicate that the preferred binding site for the water molecule is the phenolate oxygen of the anion.


