Tau Protein Binding Modes in Alzheimer’s Disease for Cationic Luminescent Ligands
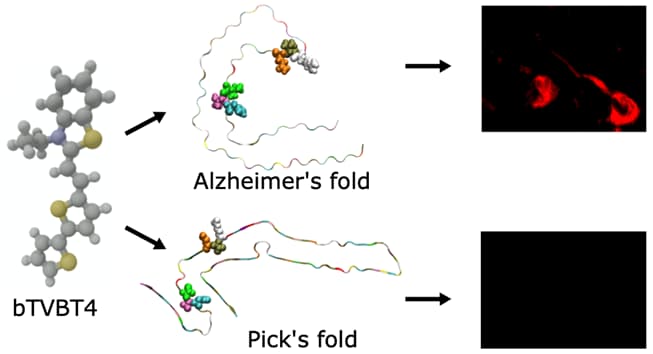
The bi-thiophene-vinylene-benzothiazole (bTVBT4) ligand developed for Alzheimer’s disease (AD) specific detection of amyloid tau has been studied by a combination of several theoretical methods and experimental spectroscopies. With reference to the cryo-EM tau structure of the tau protofilament (Nature 2017, 547, 185), a periodic model system of the fibril was created, and the interactions between this fibril and bTVBT4 were studied with non-biased molecular dynamics simulations. Several binding sites and binding modes were identified and analyzed, and results for the most prevailing fibril-site and ligand-modes are presented. Spectrum simulations were performed with a development version of the VeloxChem program into which we have implemented an automatized workflow scheme for the force-field generation and polarizable-embedding parameter derivation needed to emulate the protein environment. A key validation of the simulation work is provided by the favorable comparison of theoretical and experimental absorption spectra of bTVBT4 in solution and bound to the protein. It is conclusively shown that the ligand–protein binding occurs at the hydrophobic pocket defined by residues Ile360, Thr361, and His362. This binding site is not accessible in the Pick’s disease (PiD) fold and fluorescence imaging of bTVBT4-stained brain tissue samples from patients diagnosed with AD and PiD provides strong support for the proposed tau binding site.
Figure caption: Luminescent conjugated oilgothiphenes are developed for early-stage detection of Alzheimer’s disease. The ligand bTVBT4 shows specificity for detection of tau proteins in the Alzheimer’s fold by means of fluorescence imaging.
References:
- Todarwal, Y., Gustafsson, C., Thi Minh, N. N., Ertzgaard, I., Klingstedt, T., Ghetti, B., Vidal, R., König, C., Lindgren, M., Nilsson, K. P. R., Linares, M., Norman, P. Tau Protein Binding Modes in Alzheimer’s Disease for Cationic Luminescent Ligands. Phys. Chem. B 125, 11628 (2021); https://doi.org/10.1021/acs.jpcb.1c06019
- Rinkevicius, Z., Li, X., Vahtras, O., Ahmadzadeh, K., Brand, M., Ringholm, M., List, N. H., Scheurer, M., Scott, M., Dreuw, A., Norman, P. VeloxChem: A Python-driven density functional theory program for spectroscopy simulations in high-performance computing environments. WIREs Computational Molecular Science 10, e1457 (2020); https://doi.org/10.1002/wcms.1457


